Introduction to Qiime
Overview
The Qiime2 package is an open-source system that incorporates multiple, stand-alone programs, giving you many options to run your analysis. The different programs are provided as plug-ins to provide maximum flexibility. The Qiime2 docs webpage has all the information you need to get started on processing metabarcoding data. There are multiple tutorials available, including a detailed overview of the available plugins and links to the concepts involved.
It is possible to run the entirety of your analysis within the Qiime2 system, but today we will just show how we incorporate one of its components, taxonomy assignment, into our workflow. Today, we will just give a quick guided tour of the system. This is only a starting point, and after this you should be able to go and try any of the other tutorials on the website with your own data.
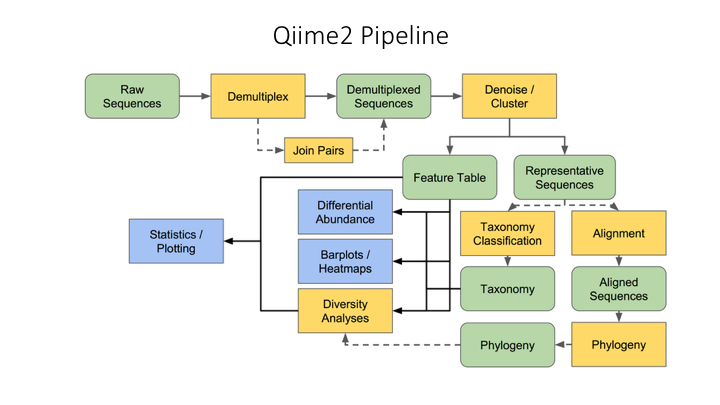
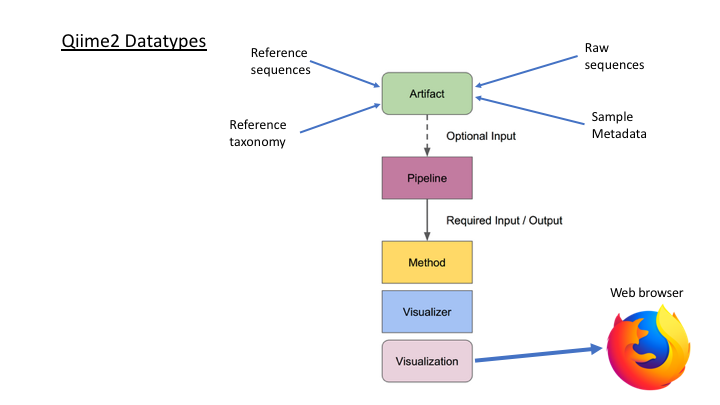
Here is an overview of the Qiime2 workflow:
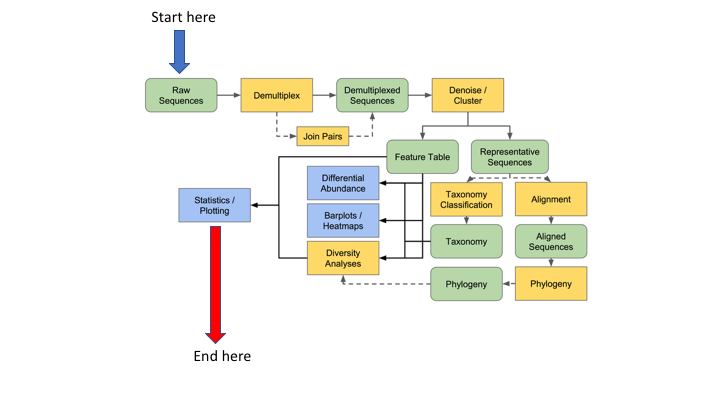
Risk of ‘copy/paste’ tutorials
Going through the tutorial, merely copying and pasting the commands:
…Has the risk of not absorbing the information as you go through:
The problem is that often you do not want to start at the beginning or finish at the end
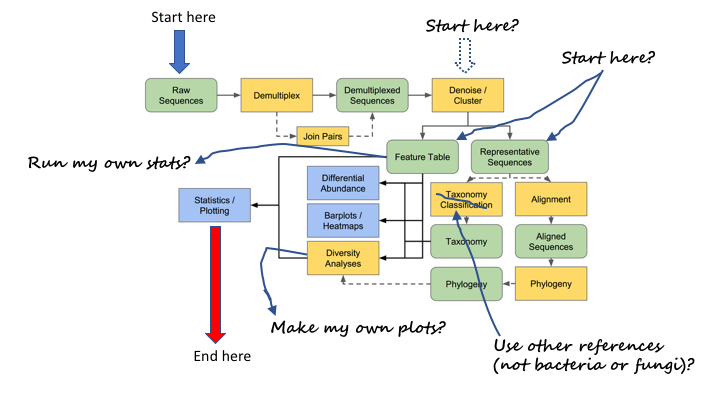
This workshop will attempt to help you work through many of the examples yourself, so you will be able to better utilise the resources.
Tour of Qiime2 website
Now we will look over the resources on the Qiime2 website

Qiime2 basics
One of the main concepts of using Qiime is that all data is represented by what they refer to as “artifacts”, which are actually compressed folders that contain the data and metadata about the data. This is done so that all data files are tracked and logged through the system. In order to use Qiime data must be imported into their system; as well, to use the outputs of Qiime in other programs they must be exported (though some visuals provide a way to download flat files of some outputs).
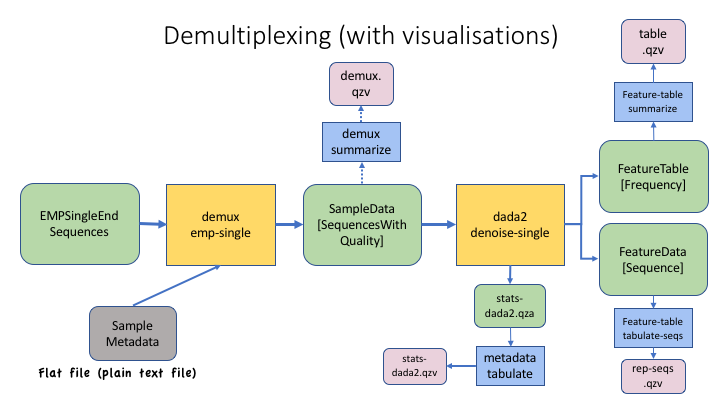
Here is an example process, with outputs and visualisations added so you can see how you can monitor the progress.
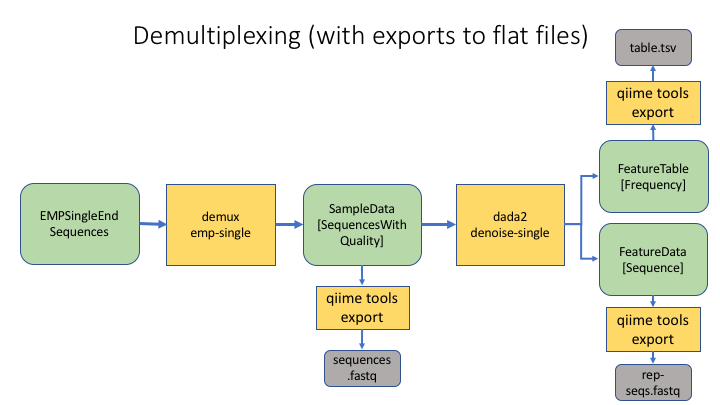
And here is the same process, with exports to flat files
Getting started: importing files into Qiime2 format
We first need to import the files produced in this morning’s lesson into the Qiime2 format so they can be processed. In the command line, we will first go to the taxonomy folder and load the Qiime2 module:
$ pwd
/home/username/edna/
$ cd taxonomy
Otherwise,
cd ~/edna/taxonomy
$ module purge
$ module load QIIME2/2021.2
To see the options for any Qiime2 command, you can use help:
$ qiime tools import --help
There are just a few arguments for importing files, but many possible types and formats. Two of the commands are sort of help commands in that they show what is possible to import.
We can view those by just entering the option:
$ qiime tools import --show-importable-types
Sometimes it is necessary to specify the format of the input file. We can view those as well:
$ qiime tools import --show-importable-formats
These two arguments provide a good guide when you are trying to figure out kind of file you are importing
Importing OTU fasta file and frequency table
Now we will write scripts to import the OTU fasta file and frequency table. Create a text file called import_otu_to_qiime.sh in the scripts folder.
In addition to the shebang on the first line, add a line to load the qiime module:
module load QIIME2/2021.2
The script will move to the otus folder to run the command:
cd ../otus
Now we will add the Qiime command. For this step, we will introduce a new way of adding the command. When we have commands with a fair number of arguments, we can split this across multiple lines. This makes it easier to read.
Type the Qiime import command like this:
qiime tools import \
--type 'FeatureData[Sequence]' \
--input-path otus.fasta \
--output-path otus.qza
You will add a backwards slash after every line (\). This tells the script to continue the command on to the next line. It is important not to have any characters, even spaces, after the backslash, or the script will think that the next line is a new command, and not a continuation of the original command. This can lead to some weird error messages, but you will learn how to fix this problem.
Once you have your script, save it and make it executable (REVIEW: chmod a+x SCRIPTNAME). Then run it in the terminal
./import_otu_to_qiime.sh
If the script worked then you should have a file called otus.qza in your /otus folder. All Qiime artifacts end in .qza, and all visuals end in .qzv.
We cannot import the frequency table directly into Qiime. First we have to convert our table into the biom format. Then the biom format will be imported to Qiime. Create a script called import_freq_table_to_qiime.sh in your scripts folder. Add the shebang, cd and qiime module lines, and then put these commands:
biom convert -i otu_frequency_table.tsv \
-o fish_frequency.biom \
--to-hdf5
qiime tools import \
--input-path fish_frequency.biom \
--type 'FeatureTable[Frequency]' \
--input-format BIOMV210Format \
--output-path otu_frequency_table.qza
The output of this script should be two files: a .biom file and a qiime .qza frequency table file.
Create a phylogeny from OTUs
There is one last script we will run before we move on to taxonomy assignment. This is to align and create a phylogenetic tree from the OTUs. This can be done with separate commands, but Qiime provides a pipeline that 1) aligns the OTUs, 2) mask uninformative or ambiguous sites in the alignment, 3) infers a phylogenetic tree from the alignment, and then 4) roots the tree at the midpoint. You can check all the options at the Plugin page. You can also run these commands separately. There are other options for phylogeny, including inferring phylogeny with the iqtree and RaXMl programs. Have a look at all the options on the main Phylogeny plugin page. These tools can come in handy for multiple applications.
qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences otus.qza \
--o-alignment aligned-rep-seqs.qza \
--o-masked-alignment masked-aligned-rep-seqs.qza \
--o-tree unrooted-tree.qza \
--o-rooted-tree rooted-tree.qza
Later we will learn how to export Qiime-formatted artifacts.